Kinetic Theory 7 – Transport Coefficients 2
Last month’s blog presented the prototype ‘algorithm’ for relating macroscopic transport properties (e.g., the diffusion coefficient) to their microscopic mechanical attributes (e.g., the mean free path). This post extends the analysis by giving an elementary expression for relating viscosity and heat conduction to the mean free path, mean speed, and related molecular terms.
Viscosity
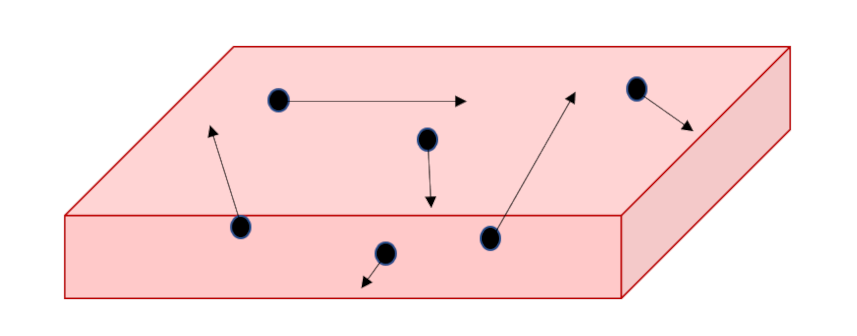
Viscosity as a macroscopic physical phenomenon has been discussed in previous blogs (here and here) and so only short summary will be provided here. The basic idea is that flow is often fixed or stagnant on a one surface while moving on another. This is the essential point that Prandtl realized in his concept of boundary layer flow. The prototypical example is the flow is shown in the figure below where some fluid is forced to flow in the $x$-direction between two plates, separated by a distance $w$ in the $y$-direction, by moving the top plate with a velocity ${\vec u} = U {\hat x}$ with respect to the bottom plate which remains fixed.
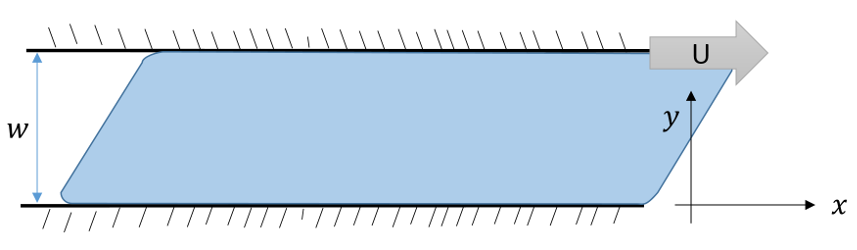
The $x$-component of the fluid’s velocity $u_x$ varies as a function of $y$ and the usual definition of the stress in the problem relates it to the velocity gradient
\[ d_{xy} = \mu \frac{\partial u_x(y)}{\partial y} \; . \]
The physical meaning of the stress is that $d_{xy}$ is the amount of momentum $p_x$ in the $x$-direction transported across any plane in the $y$-direction, which we will call $p_{xy} = – d_{xy}$ (with an appropriate change in sign to account for difference between a force acting on the fluid and the reaction of the fluid itself).
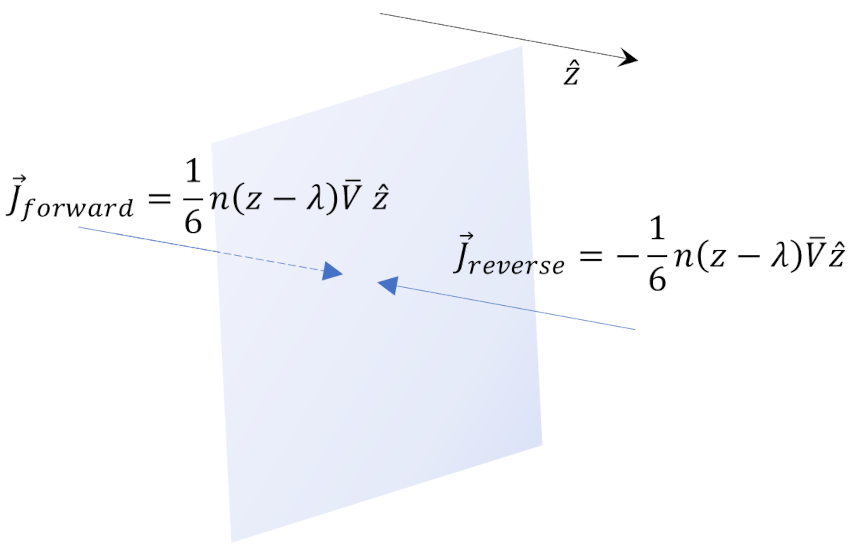
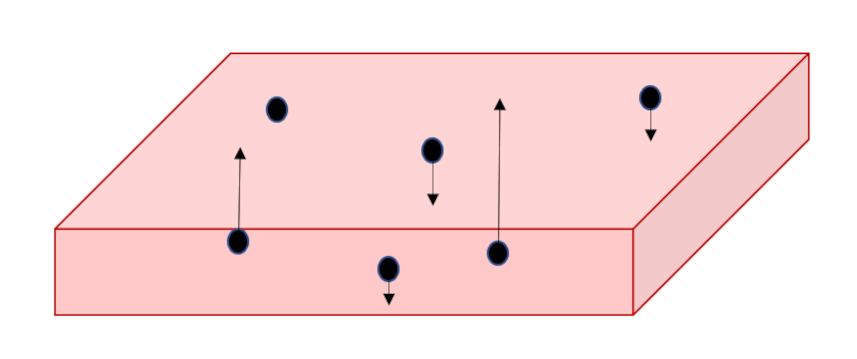
Again, using the Reif’s argument $1/6$ of the molecules will be crossing some plane $y=Y$ in the upward direction and $1/6$ of them will crossing downward. Assuming the molecular mass as $M$ and the number density as $n$, the difference in momentum is
\[ p_{xy} = \frac{1}{6} M n {\bar V} \left[ u_x(y – \lambda) – u_x(y + \lambda) \right] = – \frac{1}{3} M n \frac{\partial u_x}{\partial y} \lambda \; . \]
Comparing the two expressions gives the viscosity in terms of the microscopic parameters as
\[ \mu = \frac{1}{3} M n {\bar V} \lambda = M n D \; . \]
As with all these types of results, this one should be taken with a grain of salt regarding the numeric factor. Other authors also get this $1/3$ but they do so using quite different ways of ‘averaging’ across the populations. Nonetheless, the functional dependence of the viscosity on the microscopic parameters is correct and, in this case, the analysis of this dependence leads to some interesting conclusions.
The first thing to note is that Reif expresses the mean free path generally as
\[ \lambda = \frac{1}{\sqrt{2} \sigma n } \; , \]
where $\sigma$ is the generic cross-section, which takes the value $\pi d^2$ for hard sphere collisions giving the expression derived earlier. This generalization will prove useful in a bit but first let’s look at using this formula for the mean free path explicitly for viscosity. Putting these together gives an expression for the viscosity
\[ \mu = \frac{1}{3 \sqrt{2} \sigma} M {\bar V} \; \]
that may be quite surprising. The amount of viscosity delivered by a gas is independent of density, at least over a wide range of values for the physical parameters that enter into this theory. The limitations occur in the limits of a very small mean free path, in which the gas molecules are nearly always colliding with each other, or when the mean free path is larger than the physical size of the experiment. Kittel and Kroemer, in their book Thermal Physics, cite a quote from Robert Boyle in 1660 as reading
Before pressing on with more physics, it is worth noting that there are two satisfying things about the above quote. The first is that Boyle was scrupulous enough to report the null result of performing this experiment; a sentiment that bucks the trend of modern science where only ‘breakthroughs’ are reported. Second, it is refreshing to hear the ‘snark’ that Boyle conveys on his own behalf.
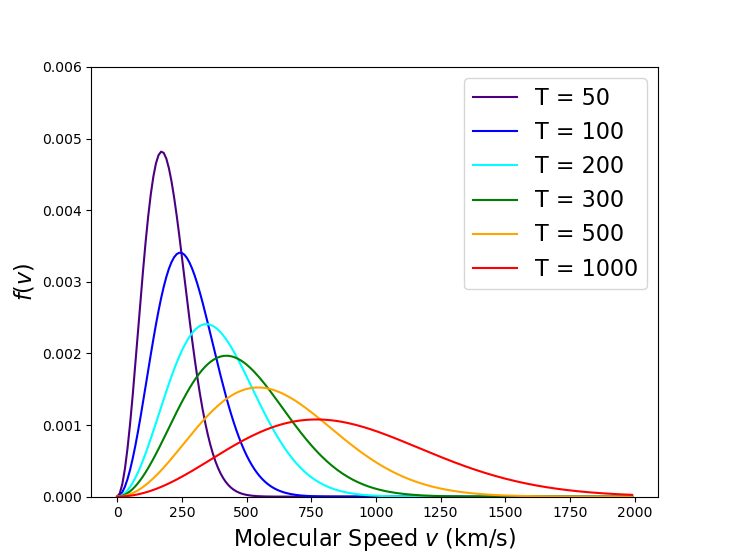
Finally, Reif presents how the viscosity changes as a function of temperature. There is an obvious dependence of speed through the explicit appearance of ${\bar V} \propto T^{1/2}$ in calculating the viscosity. And, if the collision process were accurately modeled in terms of hard spheres this would be all there was to the dependence. However, the scattering cross section is, generically, a function of speed since the underlying forces (i.e., Coulomb scattering) have a stronger influence on the particle when it is moving slowly. This is where the relaxation of the hard sphere scattering assumption becomes relevant as the scattering cross section becomes a function of speed, which, in turn, is a function of temperature in the general case. Thus, in the general case
\[ \mu = \mu \left[ {\bar V}(T), \sigma ( {\bar V}(T)) \right] \; \]
leading to an overall dependance that Reif cites going as
\[ \mu \propto T^{0.7} \; . \] An interesting implication of this functional dependence (with or without the extra, implicit temperature dependence coming through the effective cross section) is that the viscosity of a gas increases with increasing temperature, which is the exact opposite effect as is active in the vast majorities of liquids.